Visualize GSEA result with input list of gene symbols.
Source:R/AllGenerics.R, R/sig_gseaplot-methods.R
sig_gseaplot.RdVisualize GSEA result with multiple lists of genes by using clusterProfiler.
sig_gseaplot(
data,
sigs,
group_col,
target_group,
gene_id = "SYMBOL",
slot = "counts",
method = c("dotplot", "gseaplot"),
col = "-log10(p.adjust)",
size = "enrichmentScore",
pvalue_table = FALSE,
digits = 2,
rank_stat = "logFC",
...
)
# S4 method for MArrayLM,vector
sig_gseaplot(
data,
sigs,
group_col,
target_group,
gene_id = "SYMBOL",
slot = "counts",
method = c("dotplot", "gseaplot"),
col = "-log10(p.adjust)",
size = "enrichmentScore",
pvalue_table = FALSE,
digits = 2,
rank_stat = "logFC",
...
)
# S4 method for MArrayLM,list
sig_gseaplot(
data,
sigs,
group_col,
target_group,
gene_id = "SYMBOL",
slot = "counts",
method = c("dotplot", "gseaplot"),
col = "-log10(p.adjust)",
size = "enrichmentScore",
pvalue_table = FALSE,
digits = 2,
rank_stat = "logFC",
...
)
# S4 method for DGEList,ANY
sig_gseaplot(
data,
sigs,
group_col,
target_group,
gene_id = "SYMBOL",
slot = "counts",
method = c("dotplot", "gseaplot"),
col = "-log10(p.adjust)",
size = "enrichmentScore",
pvalue_table = FALSE,
digits = 2,
rank_stat = "logFC",
...
)
# S4 method for ANY,ANY
sig_gseaplot(
data,
sigs,
group_col,
target_group,
gene_id = "SYMBOL",
slot = "counts",
method = c("dotplot", "gseaplot"),
col = "-log10(p.adjust)",
size = "enrichmentScore",
pvalue_table = FALSE,
digits = 2,
rank_stat = "logFC",
...
)
# S4 method for list,ANY
sig_gseaplot(
data,
sigs,
group_col,
target_group,
gene_id = "SYMBOL",
slot = "counts",
method = c("dotplot", "gseaplot"),
col = "-log10(p.adjust)",
size = "enrichmentScore",
pvalue_table = FALSE,
digits = 2,
rank_stat = "logFC",
...
)Arguments
- data
expression data, can be matrix, DGEList, eSet, seurat, sce...
- sigs
a vector of signature (Symbols) or a list of signatures
- group_col
character or vector, specify the column name to compare in coldata
- target_group
pattern, specify the group of interest as reference
- gene_id
character, indicate the ID type of rowname of expression data's , could be one of 'ENSEMBL', 'SYMBOL', ... default 'SYMBOL'
- slot
character, indicate which slot used as expression, optional
- method
one of "gseaplot" and "dotplot", how to plot GSEA result
- col
column name of
clusterProfiler::GSEA()result, used for dot col when method = "dotplot"- size
column name of
clusterProfiler::GSEA()result, used for dot size when method = "dotplot"- pvalue_table
logical, if to add p value table if method = "gseaplot"
- digits
num, specify the number of significant digits of pvalue table
- rank_stat
character, specify which metric used to rank for GSEA, default "logFC"
- ...
params for function
get_de_table()and functionenrichplot::gseaplot2()
Value
patchwork object for all comparisons
Examples
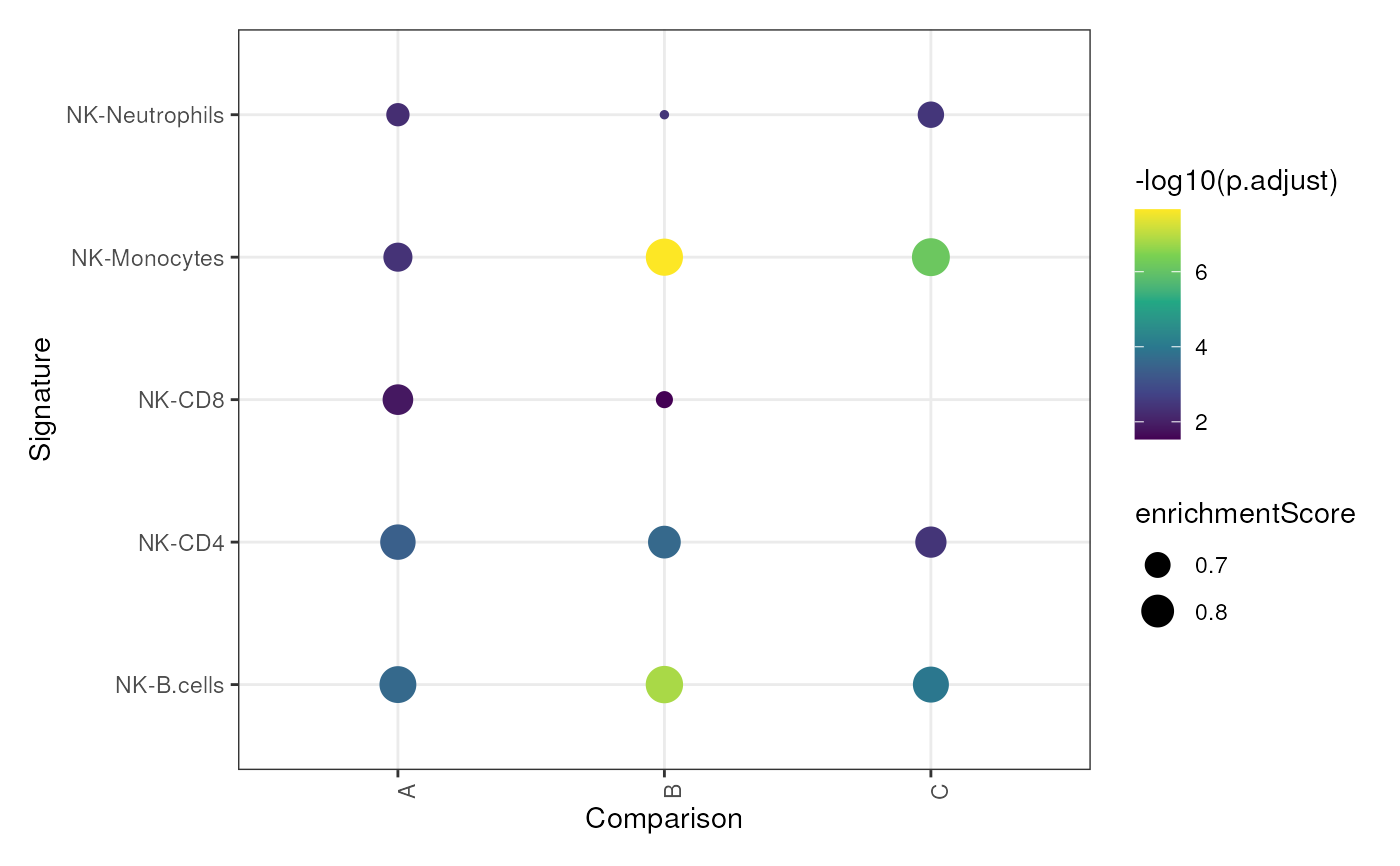
data("im_data_6", "nk_markers")
sig_gseaplot(
sigs = list(
A = nk_markers$HGNC_Symbol[1:15],
B = nk_markers$HGNC_Symbol[20:40],
C = nk_markers$HGNC_Symbol[60:75]
),
data = im_data_6, group_col = "celltype:ch1",
target_group = "NK", gene_id = "ENSEMBL"
)
#> 'select()' returned 1:many mapping between keys and columns
#> NK-Neutrophils NK-Monocytes NK-B.cells NK-CD4 NK-CD8
#> Down 4009 3946 3143 2698 2153
#> NotSig 1486 2683 4418 4991 6191
#> Up 4926 3792 2860 2732 2077
#> 'select()' returned 1:many mapping between keys and columns
#>
#> preparing geneSet collections...
#> GSEA analysis...
#> Warning: There are ties in the preranked stats (0.88% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> leading edge analysis...
#> done...
#> preparing geneSet collections...
#> GSEA analysis...
#> Warning: There are ties in the preranked stats (1.05% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> leading edge analysis...
#> done...
#> preparing geneSet collections...
#> GSEA analysis...
#> Warning: There are ties in the preranked stats (1.35% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> leading edge analysis...
#> done...
#> preparing geneSet collections...
#> GSEA analysis...
#> Warning: There are ties in the preranked stats (2.2% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> leading edge analysis...
#> done...
#> preparing geneSet collections...
#> GSEA analysis...
#> Warning: There are ties in the preranked stats (2.31% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> leading edge analysis...
#> done...